
Medical Device CE Marking & MDR Services
If you plan to market a medical device in the European Economic Area (EEA), you must comply with the European Medical Device Regulation (EU) 2017/745 (MDR). The regulation sets out strict rules to ensure devices placed on the market are safe, perform as intended, and are properly monitored.
Compliance involves more than obtaining a certificate. Manufacturers need clear and well-structured technical documentation, a sound clinical evaluation, reliable product traceability, and an effective post-market surveillance system. These obligations continue for as long as the device remains on the market.
Companies must also maintain a compliant quality management system and be prepared for ongoing oversight from Notified Bodies. Regular reviews, audits, and updates to documentation are part of maintaining conformity.
Practical regulatory guidance can support manufacturers and distributors in understanding requirements, addressing gaps, responding to Notified Body questions, and strengthening internal processes to maintain stable access to the European market.
Scope of MDR in Norway
MDR covers more than many companies initially expect. It applies to:
- Standard medical devices
- Implantable products
- Active and electronic devices
- Software with a medical purpose
- Certain aesthetic devices with medical intent
If the product meets the definition of a medical device, compliance is not optional. It must meet MDR requirements before it can be CE marked and sold.
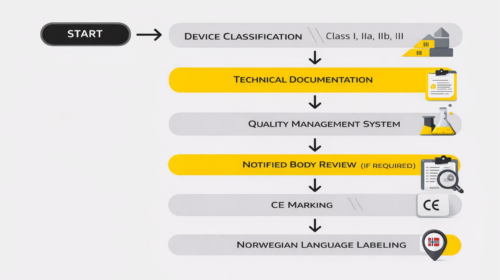
Classification of Medical Devices
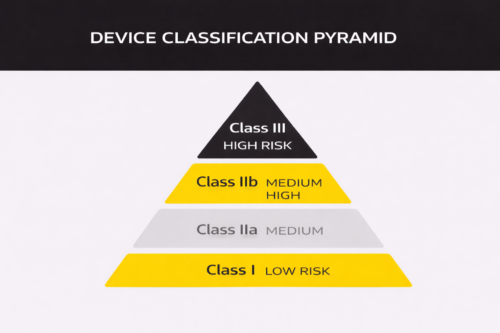
Every device must be classified according to risk. The classes range from I (lower risk) to III (highest risk). This step is not just administrative. The classification determines:
● Whether a Notified Body is involved
● How much clinical evidence is required
● The depth of review
● The expected timeliness
Incorrect classification often leads to delays, especially once the file reaches external review.
What MDR Requires in Practice
Quality Management System
A functioning Quality Management System is essential. Most manufacturers use ISO 13485 as the framework, but regulators increasingly look beyond the certificate itself. They want to see whether risk management is actively maintained. Whether complaints are actually reviewed. Whether post-market data feeds back into the system. A QMS under MDR must work in practice, not only on paper.
Technical Documentation
Technical documentation is central under MDR. It must clearly show how the device meets the General Safety and Performance Requirements. That includes design details, manufacturing information, risk files, clinical evaluation, labeling, and post-market planning. What has changed compared to previous legislation is the depth expected. Files are reviewed more critically, and inconsistencies are quickly identified.
Clinical Evaluation
Clinical evidence is one of the biggest shifts under MDR. Manufacturers must demonstrate not assume, that their device is safe and performs as intended. Literature equivalence claims are more difficult to justify than before. For higher-risk devices, clinical investigations are often required. Even for lower-risk products, the reasoning must be well documented.
Post-Market Surveillance
Under MDR, compliance continues after the product is on the market. Manufacturers are expected to actively monitor performance, analyze complaints, identify trends, and report serious incidents. For certain classifications, Periodic Safety Update Reports (PSURs) are mandatory. Authorities expect a structured, proactive system, not reactive handling.
CE Marking and Access to the Norwegian Market
To place a device on the market in Norway, the company must:
- Meet MDR requirements
- Complete the appropriate conformity assessment
- Obtain certification if required
- Affix CE marking
- Register relevant information in EUDAMED where applicable
Once CE marked under MDR, the device can circulate throughout the EEA, including Norway.
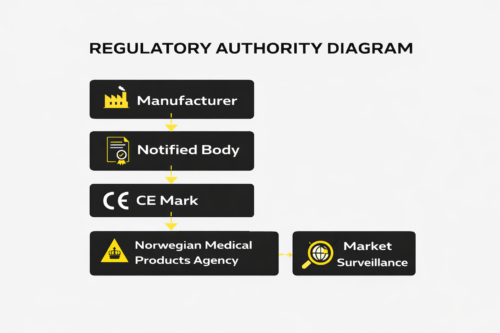
Responsibilities Across the Supply Chain
MDR clearly distributes responsibility among economic operators. Manufacturers carry primary accountability. Non-EEA manufacturers must appoint an Authorized Representative within the EEA. Importers must verify compliance before placing products on the market.
Distributors must ensure traceability and proper handling. These roles are not symbolic. Authorities increasingly assess whether responsibilities are clearly defined and documented.
EUDAMED and UDI
EUDAMED introduces a higher level of transparency across Europe. Manufacturers are required to register economic operators and upload device information. A Unique Device Identification (UDI) system must also be implemented to ensure traceability throughout the lifecycle. These measures strengthen market surveillance and incident management.
Market Surveillance in Norway
The Norwegian Medical Products Agency conducts inspections and can request technical documentation at any time.
If deficiencies are identified, companies may face corrective actions, sales restrictions, or product withdrawal. Regulatory risk today is as much about documentation readiness as it is about product safety.
Transition from MDD to MDR
Many legacy devices are still transitioning from the previous Medical Device Directive (MDD). The transition typically involves rebuilding technical documentation, expanding clinical evidence, updating labeling, and strengthening post-market systems. Given ongoing capacity constraints at Notified Bodies, early preparation remains critical.
Lifecycle-Based Compliance
MDR is built around lifecycle thinking. Certification is not the endpoint. Documentation must be maintained. Risk management must be updated. Post-market data must be reviewed continuously. Companies that treat compliance as a one-time project often struggle during inspections.
How Yallow Life Science Supports Companies
Yallow Life Science Norway works with medical device companies operating in Norway and across the EEA to strengthen regulatory foundations and maintain inspection readiness.
Support typically includes:
- MDR gap assessments
- Regulatory roadmap development
- QMS implementation and refinement
- Technical documentation restructuring
- Clinical evaluation strategy
- Post-market surveillance framework setup
- Inspection preparation
The objective is not only CE marking, but long-term regulatory stability.
Medical Device Regulations in Norway: Structured, Practical Support
Medical device compliance in Norway requires more than documentation. It requires internal alignment, traceability, and continuous oversight. Yallow Life Science supports companies seeking.